Materials Genomics
Unit 8: Regression and Generalization in Materials Data
FAU Erlangen-Nürnberg




§0 · Frame
01. Where We Are
Recap — Units 1–7 (MG) and the parallel tracks
- MG Unit 6: a material is now a feature vector — composition-only Magpie, globally-pooled RDF, local descriptors (ACSF, SOAP), pooled to a material-level \(\mathbf{x}\).
- MFML W7 (two weeks ago): bias-variance, regularization, \(k\)-fold CV, validation/test discipline — the generic ML pipeline.
- ML-PC Unit 7 (W7 lecture cancelled — public holiday; offered as self-study): process windows, robustness, hyperparameter optimization on AM/process data.
Today — MG Unit 8
- We have \(\mathbf{x}_i\) from Unit 6. We have a target \(y_i\). We can already train a regressor.
- The hard question this unit answers: what does it mean for a materials regression result to be scientifically trustworthy?
- The answer is split design, residual analysis, and reporting discipline.
02. Learning Outcomes
By the end of 90 minutes, you can:
- Apply the MFML W7 empirical-risk / regularization / CV vocabulary to materials regression problems.
- Diagnose the four materials-specific failure modes — chemistry-family leakage, polymorph aliasing, narrow-coverage bias, prototype dominance — in a regression report.
- Choose between random, chemistry-aware, structure-aware, leave-one-cluster-out, and time-based splits to match the scientific claim.
- Evaluate a formation-energy regression benchmark against the mandatory-baseline ladder and the leakage checklist.
- Read per-element, per-prototype, and per-space-group residuals to surface chemistry biases invisible to a global MAE.
- Decide from a learning curve whether a result is data-limited, model-limited, or representation-limited.
- Audit a published materials-regression paper against the MG U8 trustworthy-reporting checklist.
03. Today’s Map
Seven sections, ~90 min
- §A · MFML W7 recap, applied to materials (~10 min, 5 slides)
- §B · Materials-specific generalization failures (~22 min, 11 slides)
- §C · Split design for materials data (~15 min, 8 slides)
- §D · Formation-energy regression — the canonical case (~15 min, 8 slides)
- §E · Beyond R² — diagnostic plots (~12 min, 8 slides)
- §F · Trustworthy reporting (~8 min, 5 slides)
- §G · Wrap-up (~3 min, 2 slides)
The single sentence to leave with
In materials ML, the test set’s relationship to the training set is the scientific claim. A model is trustworthy only when its split design matches the claim its predictions are meant to support.
5-minute break after §C (around minute 47).
§A · MFML W7 Recap, Applied to Materials
04. What MFML W7 Gave Us
The empirical-risk picture
\[\hat{\boldsymbol{\theta}} \;=\; \arg\min_{\boldsymbol{\theta}} \;\underbrace{\frac{1}{N}\sum_{i=1}^{N} \ell(f_{\boldsymbol{\theta}}(\mathbf{x}_i), y_i)}_{\text{empirical risk}} \;+\; \underbrace{\lambda\,\Omega(\boldsymbol{\theta})}_{\text{regularizer}}\]
- \(\ell\) — prediction loss (squared, absolute, Huber)
- \(\Omega\) — complexity penalty (ridge, lasso, elastic-net)
- \(\lambda\) — bias-variance dial (Bishop 2006)
The materials twist
- The minimization is over the training sum.
- Generalization means the model behaves on \((\mathbf{x}, y)\) pairs not in the training sum.
- For materials data, “not in the training sum” is ambiguous — and that ambiguity is the entire scientific question of this unit.
05. The Bias-Variance Picture, Plus the Materials Term
Standard decomposition (Bishop 2006; Murphy 2012)
\[\mathbb{E}[\,(\hat{f}(x) - y)^2\,] \;=\; \underbrace{\text{Bias}^2}_{\text{model too simple}} + \underbrace{\text{Var}}_{\text{too sensitive to data}} + \underbrace{\sigma^2}_{\text{irreducible}}\]
- High bias: ridge \(\lambda\) too large, model too rigid.
- High variance: tree too deep, GNN over-parameterized.
- Irreducible noise: target is intrinsically uncertain.
The fourth term materials adds
\[+\; \underbrace{\Delta_{\text{shift}}}_{\text{distribution shift error}}\]
- Even at zero bias, zero variance, zero noise, a materials model is wrong if the training distribution differs from the deployment distribution.
- Random IID splits estimate Bias² + Var + \(\sigma^2\).
- They do not estimate \(\Delta_{\text{shift}}\).
- \(\Delta_{\text{shift}}\) is the headline content of §B.
06. Regularization as Prior, Applied to Materials Descriptors
Ridge \(\Omega(\mathbf{w}) = \|\mathbf{w}\|_2^2\) — Gaussian prior on weights.
Lasso \(\Omega(\mathbf{w}) = \|\mathbf{w}\|_1\) — Laplace prior, drives weights exactly to zero.
Elastic net — both, weighted (Bishop 2006).
These are MFML W7 content. We use them, we do not re-derive them.
Why ridge is the materials default
- Magpie has \(\sim 130\) elemental-statistics features.
- Mean atomic number, mean atomic mass, mean valence-electron count, mean ionization energy all move together along the periodic table.
- Pairwise correlations \(|r| > 0.9\) are common.
- Unregularized least squares is unstable on this design matrix.
- Ridge with \(\lambda\) chosen by 5-fold CV is the right default for any composition-only baseline.
07. Cross-Validation — Mechanically Familiar, Conceptually Wrong by Default
\(k\)-fold CV recap (Bishop 2006)
- Partition training data into \(k\) disjoint folds.
- For each fold \(j\): train on the other \(k-1\), evaluate on \(j\).
- Report mean \(\pm\) std over folds.
- Default \(k=5\) or \(k=10\).
The materials issue
- Random folds give a low-variance estimate of error on a held-out random sample.
- For most materials questions, “random sample of materials” is not the deployment distribution.
- Random-fold CV gives a low-variance estimate of the wrong quantity.
- The fix is grouped CV — folds that hold out chemistry families, prototypes, or time windows.
- This is the bridge into §B and §C.
08. The Three-Set Protocol — Same Discipline, Group-Aware Splits
The discipline (MFML W7)
- Train: fit parameters.
- Validation: select hyperparameters and model class.
- Test: evaluate the chosen model once.
If the test set influences any choice, the test estimate is no longer honest (Bishop 2006; Murphy 2012).
Materials adaptation
- All three sets must be group-disjoint in the materials-relevant grouping.
- Train and test on disjoint chemistry families and disjoint prototypes when both matter.
- Validation set used for tuning ridge \(\lambda\), GBT depth, etc. — but with the same group structure.
- Nested group CV is the gold standard when the dataset is small.
Reporting “test MAE” without specifying which axis the test set was held out along is uninterpretable. Always declare the split design.
§B · Materials-Specific Generalization Failures
09. The “What Kind of New Is Your Test Set?” Taxonomy
Six axes of “new” for a materials regression task
- New composition within a known prototype.
- New prototype within a known chemistry family.
- New chemistry family entirely.
- New database / curation source.
- New computational level (functional, \(k\)-mesh, pseudopotential).
- New measurement modality (DFT vs experiment).
The diagnostic question
Which axis or axes does your test set probe?
- A random IID split probes none of these axes.
- Different deployment scenarios demand different axes.
- Most published “test MAE” numbers are silent on this — making them uninterpretable.
10. Chemistry-Family Leakage — The Most Common Failure
The setup
- Dataset: Materials Project, all binary and ternary oxides + nitrides.
- Random 80/20 split. Train a Magpie + GBT regressor on formation energy.
- Reported test MAE: \(\sim 60\) meV/atom. Looks great.
The hidden structure
- Random split puts oxides on both sides; nitrides on both sides.
- For each test oxide, several near-neighbour training oxides exist.
- Model is doing interpolation within chemistry families, not cross-family transfer.
The reveal
- Re-split: train on oxides, test on nitrides.
- Same model, same hyperparameters.
- Test MAE on nitrides: \(\sim 250\)–400 meV/atom.
The 60-meV number was a measure of interpolation. The 250+-meV number is a measure of transfer. The model is the same; the question changed (cf. Matbench/Matbench-Discovery community benchmarks (Dunn et al. 2020; Riebesell et al. 2025)).
11. Polymorph Aliasing
The setup
- Target: bulk modulus of \(\text{TiO}_2\).
- \(\text{TiO}_2\) exists as rutile, anatase, brookite with different bulk moduli (~210, ~180, ~250 GPa).
- Composition-only descriptor: \(\mathbf{x}_{\text{rutile}} = \mathbf{x}_{\text{anatase}} = \mathbf{x}_{\text{brookite}}\).
The aliasing
- Three different \(y\) values map to one \(\mathbf{x}\) value.
- The best a composition-only model can do is predict the mean (\(\sim 213\) GPa).
- Squared-error floor is set by the polymorph variance, not by anything the model controls.
- More data does not fix this. It is a representational under-specification, not a noise problem.
Polymorph aliasing puts a noise floor on composition-only models that more data cannot remove. The fix is structural information, not statistical effort.
12. Narrow-Coverage Bias of Public DFT Databases
Materials Project, OQMD, AFLOW, NOMAD
- Cover ~\(10^5\)–\(10^6\) structures each.
- Heavily oversample: stable phases, common chemistries (oxides, halides, intermetallics), low formation energies, small unit cells.
- Heavily undersample: high-entropy alloys, disordered systems, defected phases, exotic chemistries (actinides, organic-inorganic hybrids), large unit cells.
The consequence
- A model trained on MP knows ABO₃ perovskites in detail.
- It barely knows high-entropy alloys (MP coverage: a few hundred).
- It effectively does not know defected or disordered systems.
- “Trained on Materials Project” is a much narrower claim than students usually realize.
- The training distribution is not “materials space”; it is the materials people chose to calculate.
13. OOD versus Interpolation in Descriptor Space
Two operationally different questions
- Is the test material inside the convex hull of training descriptors? → interpolation.
- Is it outside? → extrapolation / OOD.
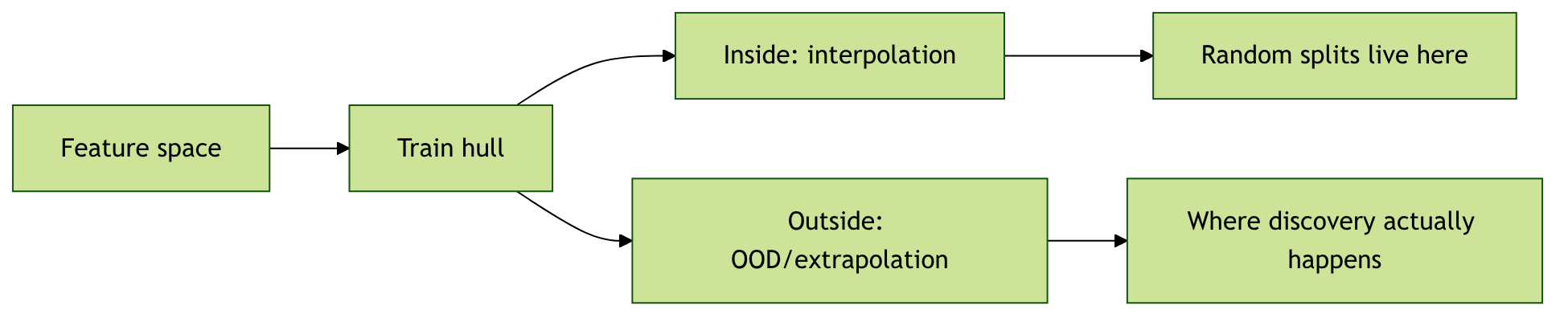
The reported-vs-relevant gap
- Most reported “good performance” numbers are interpolation numbers.
- Discovery happens at the OOD frontier, where reported performance is rarely measured.
- A simple diagnostic: train a \(k\)-NN on training descriptors. For each test point, report the minimum distance to training. Plot residuals against this distance.
- Residuals usually grow with distance; sometimes catastrophically.
14. Distribution Shift Between Databases
The same target, different numbers
- Formation energy of \(\text{Fe}_2\text{O}_3\):
- PBE in MP: \(-1.61\) eV/atom
- SCAN in OQMD subset: \(-1.68\) eV/atom
- Hybrid functional reference: \(-1.74\) eV/atom
- Differences of tens of meV/atom across databases — comparable to or larger than the best models’ MAEs.
Cross-database evaluation reveals it
- Train on MP, evaluate on the OQMD subset for the same compositions.
- Apparent test “error” \(\geq\) in-database training MAE.
- This is not model error — it is a target-definition mismatch.
- A useful materials-regression paper distinguishes between model error and database-shift error.
- In-database evaluation hides this entirely.
15. Domain Shift Between DFT and Experiment
DFT formation energies are not experimental enthalpies
- DFT misses: zero-point energy, finite-temperature contributions, configurational entropy, real-world disorder, secondary phases.
- Functional choice systematically biases binding energies (PBE: under-binds; SCAN: closer; hybrid: still imperfect).
- Direct DFT-to-experiment differences: 20–200 meV/atom, target-dependent.
The cascade
- Model trained on MP DFT formation energies.
- Used to predict candidate materials for synthesis.
- Synthesis succeeds or fails according to experimental thermodynamics, not DFT.
- A predictive MAE of 30 meV/atom against DFT is a much weaker claim than “this model can guide synthesis decisions”.
- Calibration to experimental references — when available — is essential before deployment.
16. Stability Bias and the Long Tail
Stability bias
- Public databases overrepresent stable phases (entries on the convex hull).
- Most-published research targets technologically interesting stable phases.
- The training distribution of formation energies is sharply peaked around the hull; metastable phases are underrepresented.
The deployment problem
- Discovery often needs metastable phases — kinetically accessible, technologically interesting, off-hull.
- A model trained on hull-concentrated data extrapolates poorly to metastable (high-energy-above-hull) phases.
- Reported MAEs on hull-concentrated test sets are not predictive of metastable-phase MAEs.
The materials we want to discover are often the materials we have not yet calculated. Training distributions reflect what the field has done, not what discovery requires.
17. Prototype Dominance and the Long Tail
A small set of prototypes dominates
- Rocksalt, fluorite, perovskite (ABO₃), spinel (AB₂O₄), and a handful of intermetallic prototypes account for >50% of MP entries.
- The long tail of rare prototypes — Heusler variants, layered oxides with unusual stacking, ordered defected structures — is sparsely covered.
The reporting consequence
- A model with a “good MAE” is being scored mostly on the dominant prototypes.
- Tail prototypes drive most deployment failures.
- Per-prototype residual reporting (§E) exposes this.
- A model that wins on average but loses badly on tail prototypes is not suitable for prototype-extrapolation discovery tasks.
18. Disordered, Defected, and Real Materials
Most public datasets assume idealised periodic crystals
- DFT relaxation produces a single ordered structure per entry.
- Real materials have site disorder, point defects, dislocations, grain boundaries, secondary phases, surface terminations.
- A model trained on ordered crystals applied to disordered systems is a domain shift that often goes unmeasured.
Where this bites
- High-entropy alloys: chemical disorder is the defining feature.
- Doped semiconductors: the dopant is precisely the off-ideal feature that controls properties.
- Battery cathodes during cycling: lattice deformation, vacancy ordering, phase transitions.
- ML-screening papers regularly predict ordered-DFT properties of materials whose real-world function depends on disorder. The gap is large and rarely reported.
19. The Small-Test-Set Problem
Variance of MAE estimates
- Test set size \(N_{\text{test}}\).
- Standard error on the mean MAE: \(\text{SE}(\text{MAE}) \approx \sigma_{\text{abs-resid}} / \sqrt{N_{\text{test}}}\).
- Typical materials: \(N_{\text{test}} \in [50, 5000]\).
- \(N_{\text{test}} = 100\) → SE often 5–15% of the mean.
The chemistry-coverage problem on top of that
- A single chemistry family being absent from the test set can change the mean MAE by 30%+.
- Aggregate variability of “test MAE” across reasonable test-set draws is often 30–50%.
- Reporting a point MAE without a confidence interval is statistically dishonest.
- Bootstrap or block-bootstrap CIs are the bare minimum.
§C · Split Design for Materials Data
20. Random Splits — When They Are Honest, and When They Lie
Random splits are honest when…
- “Next sample” is statistically exchangeable with training.
- The deployment task is interpolation within a chemistry/structure cluster well-represented in training.
- Examples: parameter optimization within a known polymer family; property reporting for ICSD entries similar to those in training.
…and lie when
- The deployment task involves any of the six axes from slide 09.
- The dataset has strong group structure (chemistry families, prototypes) that random sampling preserves on both sides of the split.
- The scientific claim is “discovery”, “extrapolation”, or “transfer”.
- Default to grouped splits; demote random splits to a secondary report, never a headline.
21. Chemistry-Aware Splits
Mechanics
- Hold out an entire chemistry family from training.
- Train on the rest; test on the held-out family.
- “Chemistry family” can be defined at multiple granularities:
- by anion (oxides, nitrides, halides, sulfides, …)
- by elemental presence (all materials containing Sr; all materials containing F)
- by composition cluster (Magpie-feature \(k\)-means, leave-one-cluster-out)
Implementation
sklearn.model_selection.GroupKFoldwith groups = anion family or composition cluster.- For pymatgen / matminer pipelines:
MaterialsProject.composition.alphabetical_formulareduces to the per-composition group; aggregate to family with a chemistry rule. - Concrete recipe:
groups = [classify_family(formula) for formula in train_formulae]; pass groups toGroupKFold.
The headline number this produces is the right one for cross-family transfer claims.
22. Structure-Aware and Prototype-Aware Splits
Mechanics
- Identify each material’s structural prototype.
- Hold out an entire prototype from training.
- Train on the rest; test on the held-out prototype.
Tools: - pymatgen.symmetry.analyzer.SpacegroupAnalyzer for space group and prototype matching. - AFLOW prototype labels for ICSD-derived structures. - matminer’s structure-similarity matchers for fuzzier grouping.
What this tests
- Cross-prototype transfer: does the model generalise from rocksalt to fluorite, from perovskite to spinel?
- A composition-only model performs identically on this split as on a random split — exposing its structure-blindness.
- A genuinely structure-aware model should show a smaller chemistry-vs-structure-aware gap than a composition-only one.
- This is the diagnostic for “is the structural information in the descriptor doing scientific work?”
23. Leave-One-Cluster-Out (LOCO)
Mechanics
- Cluster materials in descriptor space (e.g., \(k\)-means on standardized Magpie features), \(k \in [10, 50]\).
- For each cluster: hold it out, train on the rest, evaluate on the held-out cluster.
- Report per-cluster MAE and the worst-case cluster.
Why LOCO is informative
- Reveals which regions of materials space the model handles well.
- Worst-case cluster MAE bounds the deployment-time worst case.
- Per-cluster results identify chemistry/structure regions where the model is unreliable.
- Standard tool in molecular ML (Sheridan 2013, Kearnes et al. 2017); should be standard in materials ML too.
24. Time-Based Splits for Sequentially-Acquired Data
The setup
- Datasets that grow over time:
- Materials Project releases (2013, 2014, …, 2024, …).
- Lab notebooks accumulating new syntheses month by month.
- Autonomous-experiment platforms running 24/7.
- Train on data from \(t \leq t_{\text{cutoff}}\); test on data from \(t > t_{\text{cutoff}}\).
Why time-based is the most realistic discovery benchmark
- Mimics the deployment scenario: “would the model trained today predict tomorrow’s discoveries?”
- Captures shifts in what people calculate over time (e.g., 2013 was heavy in oxide perovskites; 2023 added more 2D materials, MOFs, high-entropy systems).
- MatBench-Discovery (Dunn et al. 2020; Riebesell et al. 2025) uses an explicit time-cutoff split.
- For active-learning loops: every iteration creates a natural train/test boundary.
25. Stratified Splits for Skewed Targets
The skew problem
- Formation-energy distributions are heavily skewed: most entries near hull, long tail of metastable.
- Band-gap distributions: many metals (band gap \(= 0\)); long tail of insulators.
- Random splits can leave the test set without high-energy / high-target examples, biasing reported MAEs downward.
Stratified splitting
- Bin the target into quantiles (typical: 5–10 quantiles).
- Sample proportionally from each bin into both train and test.
sklearn.model_selection.StratifiedKFoldfor the regression case requires manual binning then group-split.- Stratify by both target and chemistry/structure group when both matter —
StratifiedGroupKFold.
26. Split Design as Research-Question Design
The choice of split is a scientific choice
- “Predict properties of materials chemically similar to known ones” → random or stratified.
- “Predict properties of new chemistry families” → chemistry-aware.
- “Predict properties of new prototypes” → structure-aware.
- “Predict next batch our group will calculate” → time-based.
- “Predict properties broadly across materials space” → LOCO + worst-case reporting.
The protocol
- State the deployment claim explicitly.
- Choose a split design that matches the claim.
- Justify the choice in writing.
- Report headline MAE under that split.
- Report secondary MAE under random split for IID-comparison.
- Discuss the gap between them.
The split is part of the hypothesis, not the postprocessing.
27. Reporting Multiple Splits Is a Sign of Rigor
The strongest materials-regression papers report
- Random IID split: for cross-paper comparability.
- Chemistry-aware split: for cross-family transfer claims.
- Structure-aware split: for cross-prototype transfer claims.
- Time-based split: for discovery-realism claims.
- LOCO with worst-case cluster: for deployment-time worst-case bounds.
The gap is the signal
- Small gap (random vs grouped): the dataset has weak group structure, or the model is genuinely structure-aware.
- Large gap (random ≪ grouped): the dataset has strong group structure that the model exploits under random splits.
- Reporting only the smaller number is dishonest by omission.
- Reporting both, with the gap discussed, is the gold standard.
§D · Formation-Energy Regression — The Canonical Case Study
28. Why Formation Energy Is the Canonical Target
Properties that make it canonical
- Abundant: ~\(1.5 \times 10^5\) entries in MP, ~\(10^6\) in OQMD.
- Physically meaningful: \(\Delta E_f = E_{\text{compound}} - \sum_i n_i E_{\text{element}_i}\).
- Scales across chemistries: every compound has one.
- Inputs to convex-hull stability (\(E_{\text{hull}}\)) for discovery.
- Gateway to other targets: if a method cannot do formation energy, the rest of the pipeline collapses.
Order-of-magnitude reference points
- Modern GNN, MP random split: 30–40 meV/atom MAE.
- Magpie + GBT, MP random split: ~100 meV/atom MAE.
- Constant predictor (training mean): ~700 meV/atom MAE.
- Modern GNN, chemistry-aware split: 100–250 meV/atom MAE.
- “DFT vs experiment” disagreement: 50–200 meV/atom.
29. The Mandatory Baseline Ladder
Tier 0: Constant predictor
- Predict the training mean.
- MP formation energy: ~700 meV/atom MAE.
- Anything that doesn’t beat tier 0 is broken.
Tier 1: Composition-only linear
- Magpie features + ridge regression.
- ~150 meV/atom on MP random split.
Tier 2: Composition-only nonlinear
- Magpie + gradient-boosted trees (XGBoost / LightGBM).
- ~100 meV/atom on MP random split.
Tier 3: Structure-aware kernel
- SOAP descriptor + kernel ridge regression (KRR).
- ~50–80 meV/atom on MP random split.
Tier 4: Pretrained GNN
- CGCNN, MEGNet, M3GNet, EquiformerV2.
- ~30–40 meV/atom on MP random split.
Tier 5: Your method
Must clearly beat tier 4 under the split that matches your claim. Anything less has not earned its complexity.
30. What Good Performance Looks Like
Random split benchmark
- M3GNet on MP random 80/10/10 split.
- Test MAE: ~30 meV/atom.
- \(R^2\): ~0.99.
- Headline-worthy number for the IID benchmark.
Chemistry-held-out benchmark
- Same model, hold-out by anion family (test = nitrides; train = oxides + others).
- Test MAE: ~150 meV/atom.
- \(R^2\): ~0.85.
- This is the “discovery” number.
The honest report
“MAE 30 meV/atom (random split, IID-comparable to prior literature) and 150 meV/atom (chemistry-held-out, reflecting the model’s transfer-to-new-anion-family performance).”
31. Composition-Only vs Structure-Aware on This Task
Random split — surprising parity
- Magpie + GBT: ~100 meV/atom MAE.
- M3GNet: ~30 meV/atom MAE.
- ~3x advantage for the GNN.
- Many tasks: the GNN gap is much smaller under random splits because polymorphs are split across train and test.
Polymorph-rich subsets — the real GNN advantage
- Restrict to compositions with \(\geq 2\) polymorphs in MP.
- Magpie + GBT: ~150 meV/atom (hits the aliasing floor).
- M3GNet: ~50 meV/atom.
- ~3x advantage now corresponds to real structural information being used.
Structure-held-out splits
- Composition-only models perform identically on prototype-held-out as on random — they don’t see prototype.
- Structure-aware models should be penalised; if they’re not, they’re composition-only in disguise.
32. Leakage Failure Modes Seen in Published Benchmarks
Documented failures
- Test set contained polymorphs of training compositions.
- Same ICSD entry multiple times with slight symmetrization variants.
- Post-relaxation features (final volume, final stress) used as inputs to predict relaxation-derived target.
- Standard-scaler / PCA / clustering fit on full dataset before splitting.
- Time-cutoff that happens to coincide with chemistry-collection campaigns.
Inflation factors
- Each mode typically inflates headline MAE numbers by 1.5–3x.
- Stacked: a paper with two leakage modes can report 5x inflated headline performance.
- The result: a “30 meV/atom” headline that becomes “120 meV/atom” once leakage is removed.
- Many pre-2022 materials-ML headlines should be read with this in mind.
The bare-minimum check
- Train on materials with composition \(c\); test set must not contain any material with composition \(c\).
- Implement before training. Verify before reporting.
33. The Matbench Effort Riebesell et al. (2025)
What Matbench is (Dunn et al. 2020)
- Curated benchmark suite for materials-property regression / classification.
- 13 tasks: formation energy, band gap, elastic moduli, refractive index, dielectric constant, etc.
- Predefined splits — same train/test for every method.
- Per-task leaderboard at
matbench.materialsproject.org.
Why use Matbench
- IID-comparable across methods — the splits are fixed.
- Forces dedup and leakage discipline at the dataset level.
- Public leaderboard creates accountability.
- Method papers reporting Matbench numbers alongside their own splits are strictly more credible.
- Use Matbench for IID comparability; report your own group-aware splits for scientific claims.
34. The Benchmark-Choice Dilemma
Random splits — the easy trap
- Easy to implement.
- Reward interpolation.
- Cross-paper comparable.
- Hide chemistry-family leakage and prototype dominance.
- Encourage architectural over-engineering.
Chemistry-aware splits — the hard truth
- Hard to implement (requires chemistry rules).
- Reward extrapolation.
- Less cross-paper comparable (split definitions vary).
- Reveal failure modes.
- Encourage simpler, more transferable models.
The honest choice
Match the split to the abstract’s claim. If the abstract says “discovery”, the split must enforce extrapolation. If the abstract says “interpolation”, a random split is fine.
35. What Makes a Benchmark Mean Something
The five-criterion checklist
- Split design matches the deployment claim.
- Strong simple baselines (constant, Magpie+linear, Magpie+GBT) accompany the headline.
- Per-region residuals (chemistry family, prototype) reported.
- Test-set construction documented (dedup procedure, polymorph handling, leakage audits).
- Confidence intervals on the headline MAE.
The harsh reality
- Most published materials-ML benchmarks satisfy 2/5 of these.
- A benchmark that satisfies 4/5 is currently rare.
- A benchmark that satisfies 5/5 is currently exemplary.
- This is the bar the cohort should hold their own work to.
Closing of §D
Formation-energy regression is the canonical case because every failure mode shows up here clearly. Mastering this case generalises to every other materials-regression task you will encounter.
§E · Beyond R² — Diagnostic Plots for Materials
36. Residuals vs Target Magnitude
The plot
- \(x\)-axis: \(y_{\text{true}}\) (formation energy in eV/atom).
- \(y\)-axis: residual \(r = y_{\text{true}} - \hat{y}\).
- Each test material is one point.
Reading the plot
- Flat band of residuals → homoscedastic; model is uniformly accurate across the range.
- Fanning out at the tails → heteroscedastic; model is reliable in the middle, unreliable at the extremes.
- Systematic slope → calibration error; model is biased. Linear-corrected predictions usually fix this.
- For formation energies, the tails are often metastable phases — exactly where discovery happens.
37. Residuals by Composition — Per-Element Errors
The recipe
- For each element \(E\):
- \(\mathcal{T}_E = \{\text{test materials containing } E\}\).
- Compute \(\text{MAE}_E = \frac{1}{|\mathcal{T}_E|} \sum_{i \in \mathcal{T}_E} |r_i|\).
- Also: \(N_E = |\mathcal{T}_E|\), \(\bar{r}_E\) (signed mean for bias).
- Tabulate: element, \(N_{\text{train}}\), \(N_{\text{test}}\), \(\text{MAE}_E\), \(\bar{r}_E\).
Reading the table
- High \(\text{MAE}_E\) at low \(N_{\text{train}}\) → coverage limitation. More data on this element would help.
- High \(\text{MAE}_E\) at high \(N_{\text{train}}\) → physics limitation. Element’s chemistry is intrinsically harder.
- Large \(|\bar{r}_E|\) → systematic bias; model under- or over-predicts compounds containing \(E\).
- Common pattern: lanthanides and actinides have high \(\text{MAE}_E\) at low \(N_{\text{train}}\) (coverage); transition metals have moderate \(\text{MAE}_E\) at high \(N_{\text{train}}\) (physics).
38. Per-Prototype Error Tables
The recipe
- Identify each material’s prototype (
pymatgen.StructureMatcheror AFLOW prototype labels). - For each prototype \(P\):
- \(\text{MAE}_P\), \(N_P\), \(\bar{r}_P\) (per-prototype MAE, count, signed bias).
- Sort by MAE; report top-5 worst and top-5 best prototypes.
What this reveals
- Whether the model’s accuracy is uniform across structural motifs.
- Which prototypes are inherently hard (rare, complex, low-coverage).
- Which prototypes are inherently easy (common, simple, high-coverage).
- Often: cubic high-symmetry prototypes (rocksalt, fluorite) → low MAE; low-symmetry / large-unit-cell prototypes → high MAE.
The composition-only diagnostic
A composition-only model has identical per-prototype MAE under random splits as overall (modulo sample-size noise). A structure-aware model that genuinely uses structure should show prototype variation.
39. Residuals vs Space Group
The recipe
- Group materials by space group (1-230).
- Often pool by crystal system: cubic, tetragonal, orthorhombic, hexagonal/trigonal, monoclinic, triclinic.
- Per-system MAE table.
Reading the pattern
- Cubic / hexagonal: usually best — high symmetry, smaller unit cells, more training data per system.
- Monoclinic / triclinic: usually worst — low symmetry, larger unit cells, often-rare prototypes.
- Discrepancy reveals symmetry-bias in features or model.
- A model that handles all crystal systems comparably is more transferable than one strongly biased toward cubic.
40. Learning Curves — Diagnosing Data vs Model Limitation
The plot
- \(x\)-axis: \(\log N_{\text{train}}\) (e.g., \(N \in \{100, 300, 1000, 3000, 10000, 30000\}\)).
- \(y\)-axis: MAE on a fixed validation set.
- Curves: model’s training MAE; model’s validation MAE.
Three regimes
- Steep descent on validation → data-limited. More data helps; collect more.
- Flat at non-zero floor → model- or representation-limited. More data doesn’t help; change architecture or features.
- Train and val both high → target is too noisy or representation too weak.
The materials adaptation
- Compute learning curves under both random and chemistry-aware splits.
- They often disagree about which regime you are in.
- Random-split learning curve might say “model-limited”; chemistry-aware curve says “data-limited” — i.e., we have plenty of in-family data, not enough cross-family data.
41. Heteroscedastic Noise Across the Periodic Table
Not all chemistry has equal target noise
- DFT formation-energy uncertainty depends on chemistry:
- Strongly correlated electrons (transition-metal oxides with localised d-states): Hubbard-U sensitivity → 30-100 meV/atom uncertainty.
- Magnetic systems (Fe, Co, Ni compounds): magnetic ordering uncertainty.
- Lanthanides / actinides (\(f\)-electrons): functional-choice sensitivity → 100+ meV/atom.
- Light-element compounds (carbides, borides): often well-predicted.
The reporting consequence
- A model with global MAE 50 meV/atom isn’t wrong on lanthanides if the target itself has 100 meV/atom uncertainty there.
- Per-region noise estimates contextualise per-region MAE.
- A “good” model achieves MAE comparable to the irreducible target noise — and not much better.
- Reporting target-noise estimates alongside MAE is exemplary practice.
42. Calibration vs Ranking — Different Metrics, Different Use Cases
Calibration metrics
- MAE, RMSE, \(R^2\), residuals-vs-true plots.
- Question: “Is the predicted value close to the true value?”
- Useful when: reporting properties, screening with a threshold, downstream physical reasoning.
Ranking metrics
- Spearman rank correlation \(\rho\), Kendall \(\tau\), top-\(k\) recall.
- Question: “Does the model order materials correctly, even if the absolute values are off?”
- Useful when: pure screening (only the rank matters); active learning (acquisition functions use ranks).
A model can be well-ranked but poorly calibrated — useful for screening, not for property reporting. Choose the metric to match the downstream use.
43. Reading a Diagnostic Plot Suite — A Checklist
For any materials-regression paper you are reading
- Look for: residuals-vs-target plot. Read for heteroscedasticity.
- Look for: per-element error table. Read for chemistry bias.
- Look for: per-prototype error table. Read for structural bias.
- Look for: learning curves under multiple split designs. Read for data-vs-model limitation.
If they’re not there
- The paper hasn’t done the diagnostic work.
- The headline MAE is uninterpretable beyond the IID-comparison context.
- For deployment-relevant decisions, consider the paper’s claims unsupported until the diagnostics appear in a follow-up or in your own re-evaluation.
Diagnostic plots are not an aesthetic preference. They are how the field’s headline numbers acquire scientific meaning.
§F · Trustworthy Reporting
44. The Mandatory-Ablation Test for Structure-Awareness
The ablation
- Take your “structure-aware” model.
- Randomize the atomic positions of every input structure (preserve the species list).
- Re-train. Re-evaluate.
- If MAE doesn’t change much: your model is composition-only in disguise.
Why this matters
- Many published “GNN” results don’t fail this test as cleanly as the architecture diagram suggests.
- A model that uses positional information should break when positions are randomised.
- A model that doesn’t break is doing composition-only work with extra parameters.
- Mandatory for any paper claiming structure-awareness.
Variants of the ablation
- Randomise species (preserve positions): tests whether the model cares about chemistry.
- Permute species: similar.
- Both at once: confirms the model is doing nothing meaningful with structure.
45. The Mandatory Baselines
Every materials-regression paper should include
- Constant predictor (training mean).
- Magpie + linear/ridge.
- Magpie + GBT.
- Cost: a few CPU-hours total.
- Value: anchors any complexity claim against a clear “you must beat this much” reference.
Why each tier
- Constant: catches gross bugs (model achieving worse than mean — broken).
- Magpie + linear: tests whether composition contains a linear signal.
- Magpie + GBT: a strong nonlinear composition-only baseline.
The “scientific cost” of skipping baselines
- Without baselines, the reader cannot judge whether your method’s MAE is impressive.
- “30 meV/atom” might be state-of-the-art (if Magpie+GBT gets 100) or unimpressive (if Magpie+GBT gets 32).
- Without baselines, the headline number is uncalibrated.
46. Honesty About Test-Set Construction
Document everything
- Source database snapshot date (MP releases monthly; results are date-specific).
- Deduplication procedure (by formula? by structure?
StructureMatcherwith what tolerances?). - Polymorph handling (kept all? dedup to lowest-energy? randomly chose one?).
- Was the test set held out before model selection?
- Did any hyperparameter touch the test set?
Why this matters
- Two papers with the same headline MAE but different deduplication strategies are not comparable.
- A test set that wasn’t held out before model selection isn’t actually a test set; it’s a validation set.
- Most “test MAE” gaps between papers are construction-artifact gaps, not method gaps.
- Documentation lets the reader reproduce the test set verbatim.
If your reader cannot reconstruct your test set from the methods section, your headline number is unreplicable.
47. The MG U8 Trustworthy-Reporting Checklist
Before claiming a regression result
- Split design declared and matched to the deployment claim.
- Mandatory baselines present: constant, Magpie+linear, Magpie+GBT.
- Per-region residuals reported: per-element and per-prototype tables.
- Structure-awareness ablation present (if claiming structure-awareness).
- Leakage paths audited: dedup, post-relaxation features, full-data preprocessing.
- Confidence intervals on the headline MAE.
- Test-set construction documented (snapshot, dedup, holdout discipline).
Self-assessment for your own work
- 7/7: exemplary; aim here.
- 5–6/7: rigorous; current state of the best papers.
- 3–4/7: typical; most published work.
- 0–2/7: not yet ready to claim a result.
The cohort exercise targets 5/7. Your thesis work should target 7/7.
48. Anti-Patterns to Call Out by Name
The five most-common reporting failures
- Random-split numbers in a “discovery” paper.
- No baselines, or baselines that are weaker than necessary.
- Mean MAE without per-region breakdown.
- Quiet “outlier removal” — often the hardest, most informative cases.
- Hyperparameters tuned on test set, then reported as test performance.
The cultural fix
- Materials ML is a young field; peer-review norms are still maturing.
- Each cohort that reports rigorously raises the local bar.
- Each cohort member who refuses to skip the diagnostic plots in their own work and asks for them in their reading raises the field’s bar.
- This unit is part of the fix. You are part of the fix.
Trustworthy reporting is a cultural practice, not just a technical one. Build it into your habits this semester.
§G · Wrap-Up
49. Summary — Top Takeaways
The seven exam-ready statements
- Random IID splits in materials ML usually overestimate generalization. The fix is grouped splits matched to the deployment claim.
- The six axes of “new” — composition, prototype, chemistry family, database, computational level, measurement modality — diagnose what kind of generalization a test set probes.
- Polymorph aliasing puts a noise floor on composition-only models that more data cannot remove. The fix is structural information.
- Public DFT databases are what people calculated, not random samples of materials space. Training distributions inherit this bias.
- Formation-energy regression is the canonical case; the mandatory baseline ladder (constant, Magpie+linear, Magpie+GBT, structure-aware kernel, pretrained GNN) anchors any complexity claim.
- Per-element / per-prototype / per-space-group residuals reveal chemistry biases invisible to a global MAE.
- A “structure-aware” model that survives positional randomization is composition-only in disguise.
The single sentence for the exam
In materials ML, the test set’s relationship to the training set is the scientific claim. A model is trustworthy only when its split design matches the claim its predictions are meant to support.
50. Bridge to Unit 9 — Neural Networks for Materials Properties
Unit 9 (next week)
- Neural surrogates for materials properties: SchNet, CGCNN, MEGNet, M3GNet, equivariant successors (NequIP, MACE, Allegro, EquiformerV2).
- Architecture choices: graph construction, equivariance, message-passing depth, readout pooling.
- Pretrained foundation models (MACE-MP-0, MatterSim) and how to fine-tune them on small custom datasets.
What carries forward unchanged from today
- The split-design discipline of §C.
- The baseline ladder of §D2 — including the Magpie+GBT skeptic’s baseline.
- The per-region residual analysis of §E.
- The seven-point trustworthy-reporting checklist of §F4.
- A neural model is interesting only if it improves performance under a credible chemistry- or structure-aware split, with residuals that look reasonable in the chemistry families that matter.
Better architecture does not fix bad benchmarking. The MG U8 discipline carries into U9 and every subsequent unit of MG.
Continue
- ← Previous: Unit 07 — Graph-Based Crystal Representations
- → Next: Unit 09 — Neural Networks for Materials Properties
- All courses
![]()
© Philipp Pelz - Materials Genomics